
Sindromul Turner
Date generale. Sindromul Turner este o boală cromosomică determinată de monosomia completă sau parţială a cromosomului X. Sindromul Turner este una din cele mai frecvente boli cromosomice, afectând aproximativ 1/5.000 de nou-născuţi, adică circa 1/2.500 de fete.
Genetică. Sindromul Turner este o boală cromosomică determinată de absenţa completă sau parţială a unui gonosom, în condiţiile în care cel de-al doilea gonosom este cromosomul X. În cazul monosomiilor X complete, 50-70% au origine paternă, fiind consecinţa pierderii cromosomului sexual în meioza (diviziunea ce asigură formarea celulelor sexuale) paternă.
Diagnostic.
Semne clinice evidente la naştere sunt: copil de sex feminin cu lungime mai mică decât normal, limfedem (acumulare subcutanată de limfă) dur, nedureros şi tranzitoriu la nivelul mâinilor şi picioarelor, gât scurt, cu exces de piele pe ceafă şi/sau pterygium coli (pliu cutanat pe feţele laterale ale gâtului) şi distanţă intermamelonară mare.
Diagnosticul clinic în perioada copilăriei se bazează pe identificarea unui deficit major de creştere, înălţimea copilului fiind mult mai mică decât valoarea medie corespunzătoare vârstei biologice.
După vârsta pubertăţii (11-13 ani la fete) diagnosticul clinic de sindrom Turner este sugerat de următoarele semne clinice: hipostatură (înălţime mai mică de 145 cm) semne de disgenezie ovariană – amenoree primară (absenţa ciclurilor menstruale), sterilitate primară şi definitivă (ovarele nu produc ovule), glande mamare puţin dezvoltate, pilozitate axilară absentă, pilozitate pubiană redusă, organe genitale externe cu aspect infantil, linia joasă de inserţie a părului pe ceafă, terminată în trident, umeri mai laţi decât şoldurile, cubitus valgus (deformare spre exterior a antebraţului în raport cu braţul), unghii convexe hipoplazice, numeroase aluniţe, uneori malformaţii congenitale cardiace sau renale.
Confirmarea diagnosticului clinic implică analiza cromosomică şi dozări hormonale.
Examenul cromosomic este esenţial pentru stabilirea diagnosticului de certitudine. Cariotipul poate releva:
- monosomie X completă omogenă – 45,X (65% din cazuri)
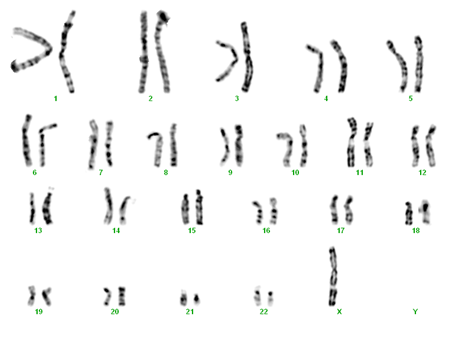
monosomie X omogenă
- monosomie X în mozaic – 45,X/46,XX sau 45,X/46,XX/47,XXX (12% din cazuri)
- monosomie X parţială prin deleţie pe cromosomul X – 46,X,del(X) (6% din cazuri)
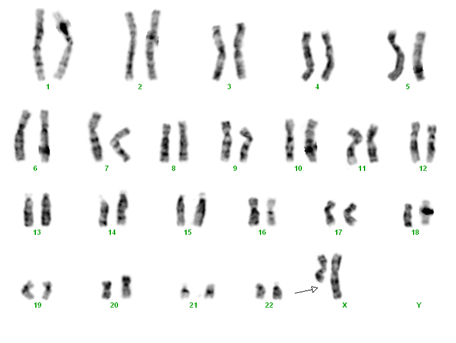
monosomie X parţială prin deleţie pe braţul lung al cromosomului X
- monosomie X parţială prin isocromosom X – 46,X,i(X) (11% din cazuri)
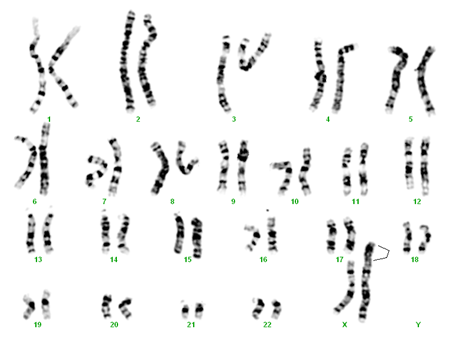
monosomie X parţială prin isocromosom X de braţ lung
- monosomie X parţială prin cromosom inelar – 46,X,r(X) (6% din cazuri).
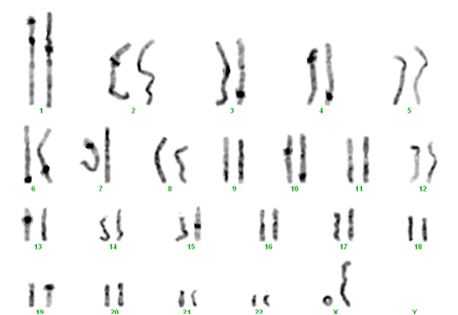
Analizele hormonale utile pentru confirmarea diagnosticului de sindrom Turner sunt: estrogeni şi progesteron – scăzute, gonadotrofine hipofizare (FSH şi LH) – nivel crescut.
Tratament. Schemele de tratement în ST sunt diferenţiate în funcţie de vârstă şi de eventualele complicaţii. Principalele elemente clinice vizate de tratament sunt: hipostatura şi deficitul de feminizare.
Corecţia deficitului de creştere este efectuată în trei intervale de vârstă:
- în timpul copilăriei (2-10 ani) este utilă administrarea de supradoze de GH (hormon de creştere); tratamentul trebuie iniţiat cât mai devreme posibil şi să fie continuu; terapia cu GH nu mai este utilă după 10 ani când receptorii pentru acest hormon nu mai răspund la supradoze;
- după vârsta de 11 ani (vârsta de debut a pubertăţii la fete) se aplică timp de 1 an – 1 an şi jumătate o terapie cu oxandrolon (un hormon sexual masculin slab) care stimulează creşterea pe lungime a oaselor;
- după vârsta de 12-13 ani se aplică terapie substitutivă cu hormoni sexuali feminini (preparate ce conţin estrogeni şi progesteron); acest tratament se face cu pilule contraceptive (conţin dozele optime de estrogeni şi progesteron) – dar nu cu scopul de preveni apariţia unei sarcini – fiind preferabilă administrarea de preparate microdozate, bazate pe hormoni naturali.
După vârsta normală de debut a pubertăţii, se face corecţia deficitului de feminizare prin administrare de amestecuri de estrogeni şi progesteron (pilule contraceptive). Această terapie trebuie continuată până în apropierea vârstei normale de instalare a menopauzei (40-50 ani).
După oprirea tratamentului cu preparate estro-progestative trebuie corectată osteoporoza care este mult mai severă decât la femeile normale, deoarece ovarele pacientelor cu sindrom Turner nu au funcţionat corect niciodată pe parcursul vieţii.
Prognosticul. În majoritatea cazurilor, evoluţia pacientelor cu sindrom Turner este favorabilă. Astfel, speranţa de viaţă este cvasinormală, exceptând situaţiile în care există anomalii congenitale viscerale cardiace sau renale. În perioada adultă pacientele cu sindrom Turner pot dezvolta hipertensiune arterială, obezitate, diabet zaharat, tiroidite Hashimoto (autoimune), cataractă, osteoporoză.
Inteligenţa este normală, cu o scădere a percepţiei spaţiale şi a capacităţii de abstractizere, astfel încât integrarea socială a pacientelor este în majoritatea cazurilor bună. În condiţiile aplicării de terapii adecvate, hipostatura şi deficitul de feminizare pot fi corectate.
Singurul aspect clinic care nu poate fi corijat este sterilitatea, în cazul femeilor cu sindrom Turner fiind imposibilă apariţia de sarcini plecând de la ovule proprii.
Profilaxie. În condiţiile în care sindromul Turner determină sterilitate, boala nu se poate transmite la descendenţi, astfel încât riscul de recurenţă a bolii la descendenţii unei paciente cu sindrom Turner este zero.
La cuplurile care au un copil cu sindrom Turner, riscul de a avea un alt copil afectat este de obicei nesemnificativ (mai mic de 1%) dar este indicat un consult genetic înainte de o nouă sarcină, existând posibilităţi de diagnostic prenatal.
Screeningul prenatal al boli se bazează doar pe evaluarea ecografică, deoarece nu există metode biochimice adecvate. Modificările ecografice sugestive pentru monosomia X sunt: hygroma chistică (acumulare în exces de limfă la nivelul cefei) şi anomalii congenitale de cord. Confirmarea prezenţei anomaliei cromosomice impune puncţie de vilozităţi corionice sau puncţie amniotică, urmată de efectuarea analizei cromosomice prin tehnici clasice sau prin metoda FISH pentru detecţia prenatală a aneuploidiilor.
Prof.dr. Eusebiu Vlad Gorduza