
Sindromul Prader-Willi
Date generale. Sindromul Prader-Willi, determinat de absenţa unor gene localizate 15q11-q13, are o incidenţă de 1/10.000-1/15.000 de nou-născuţi, majoritatea cazurilor fiind sporadice.
Genetică. În sindromul Prader-Willi există mai multe categorii de modificări genetice, dar cele mai frecvente sunt:
- deleţie 15q11-q13 de novo – prezente în 75% din cazuri (întotdeauna lipseşte fragmentul 15q11-q13 de cromosomul 15 patern), fiind caracterizate prin absenţa unui segment cromosomic de aproximativ 3-4 Mb (3-4 milioane de baze azotate);
- disomia uniparentală maternă a cromosomului 15 prezentă în 20% din cazuri (prezenţa a doi cromosomi 15, dar amândoi proveniţi de la mamă, cromosomul 15 de origine paternă lipsind);
Diagnostic. Diagnosticul clinic al sindromului Prader-Willi se bazează pe asocierea următoarelor semne clinice mai frecvente: hipotonie neonatală, probleme de alimentare în perioada de nou-născut şi sugar, hipostatură, retard mental, dismorfie cranio-facială caracteristică (îngustare temporală, ochi cu formă de migdală, gură cu colţuri căzute), obezitate după vârsta de 2-3 ani, hipogonadism (dezvoltare redusă a organelor genitale externe) şi tulburari de comportament.
Alte semne clinice mai rar întâlnite sunt: tulburări de somn, hipopigmentarea pielii şi părului (piele palidă şi păr decolorat), mâini şi picioare mici (acromicrie), miopie şi strabism, tulburări de vorbire, tulburări de comportament (încăpăţânare extremă, tendinţă la autociupire) sensibilitate dureroasă redusă.
Diagnosticul paraclinic al sindromului Prader-Willi necesită aplicarea mai multor metode.
Testarea se începe prin realizarea analizei cromosomice şi a testului FISH, folosind sonde specifice regiunii 15q11-13. Dacă aceste analize nu confirmă suspiciunea clinică, se impune efectuarea analizei MLPA pentru verificarea anomaliilor de metilare ale ADN-ului, generate de prezenţa disomiei 15 uniparentale materne.
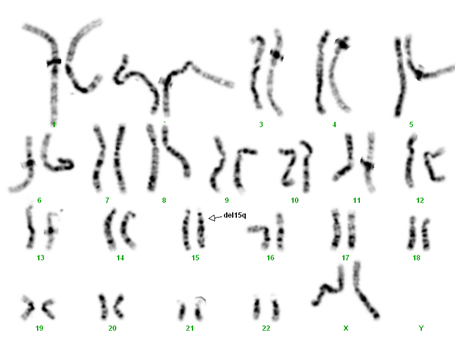
Deleţie 15q11-13
Tratament. Tratamentul este diferenţiat în diverse etape ale vieţii.
În perioada neonatală trebuie asigurat necesarul alimentar prin tehnici speciale de alăptare sau gavaj, astfel încât să nu survină un retard de creştere secundar dificultăţilor de alimentaţie asociate hipotoniei musculare extreme.
În perioada copilăriei, este utilă administrarea de hormon de creştere, care are două efecte benefice: combate hipostatura caracteristică bolii şi stimulează anabolismul ce limitează efectele negative ale hiperfagiei.
În perioada de adult este util controlul obezităţii prin stimularea exerciţiilor fizice şi administrarea de inhibitori ai serotoninei pentru a ameliora anomaliile de comportament.
Prognosticul. Speranţa de viaţă este normală, dar în perioada adultă pot exista numeroase complicaţii induse de obezitate. Unii pacienţi prezintă retard mental, dar prin stimulare adecvată în copilărie poate fi limitată severitatea acestuia. Prognosticul reproductiv poate fi marcat de existenţa hipogonadismului.
Profilaxie. Diagnosticul prenatal al afecţiunii este obligatoriu în cazurile familiale, fiind bazat pe analiza cromosomică a amniocitelor sau celulelor vilozitare şi pe teste moleculare, care permit identificarea originii parentale a cromosomilor 15. Riscul de recurenţă al bolii este diferit în raport cu tipul anomaliei. În disomiile uniparentale şi deleţiile de novo, părinţii unui copil bolnav au un risc nesemnificativ. În deleţiile moştenite riscul de recurenţă este de aproximativ 12%.
Pacienţii cu sindrom Prader-Willi au rareori descendenţi, dar riscul de a avea un copil bolnav cu sindrom Prader-Willi (dacă pacientul este bărbat) sau sindrom Angelman (dacă pacientul este femeie) este de 50%.
Prof.dr. Eusebiu Vlad Gorduza