
Sindromul Klinefelter
Sindromul Klinefelter este o boală genetică determinată de trisomia XXY, caracterizată prin prezenţa suplimentară a unuia (mai rar a mai multor) cromosomi X la o persoană de sex masculin.
Boala este cea mai frecventă anomalie gonosomală la sexul masculin, afectând aproximativ 1/1000 din nou-născuţii de sex masculin.
Genetică. Sindromul Klinefelter este o boală cromosomică determinată de prezenţa suplimentară a unui gonosom X, în condiţiile în care sunt prezenţi alţi doi gonosomi normali: unul X şi unul Y. În trisomiile XXY omogene, în majoritatea cazurilor cromosomul X suplimentar are origine maternă, existând o corelaţie statistică între riscul de sindrom Klinefelter la copil şi vârsta maternă avansată în momentul concepţiei.
Mecanism patogenic. Particularităţile clinice ale sindromului Klinefelter sunt determinate de prezenţa suplimentară a cel puţin unui cromosom X. Prezenţa în exces a unora din genele de pe cromosomul X determină anomalii în formarea testiculilor (disgenezie şi atrofie testiculară) cu lipsa secreţiei de testosteron (hormonul sexual masculin) şi absenţa producerii de spermatozoizi (azoospermie) cu sterilitate masculină primară şi definitivă.
Diagnostic.
Boala nu are semne clinice particulare în timpul copilăriei, ea fiind diagnosticată de obicei la adolescenţă datorită întârzierii dezvoltării caracterelor sexuale secundare masculine.
Principalele semne clinice evidente după pubertate sunt: statura înaltă, asocierea între microorhidie (dimensiuni reduse ale testiculilor) şi penis normal (disociaţie peno-orhitică), sterilitate masculină şi inadaptabilitate socială.
Majoritatea pacienţilor cu trisomie XXY prezintă o sexualizare masculină deficitară, determinată de deficitul de secreţie al testosteronului. Astfel, caracterele sexuale secundare sunt slab dezvoltate: pilozitate facială (barbă şi mustaţă), axilară (la subsuoară) şi tronculară (trunchi şi spate) absente sau slab reprezentate, pilozitate pubiană redusă cu aspect ginoid (feminin), corp cu conformaţie de tip feminină (şolduri mai late decât umerii), voce înaltă, depozitele de grăsime cu dispoziţie ginoidă. De asemenea, disgenezia testiculară induce azoospermie şi sterilitate primară şi definitivă. O altă particularitate fenotipică este prezenţa ginecomastiei (dezvoltarea glandelor mamare la un individ de sex masculin).
Dezvoltarea intelectuală este aproape normală, dar pacienţii cu sindrom Klinefelter prezintă tulburări de învăţare, determinate de dislexie (incapacitatea de a vorbi corect). În schimb, la pacienţii cu polisomii XY, prezenţa unui număr crescut de cromosomi X se asociază cu retard intelectual. Personalitatea pacienţilor cu sindrom Klinefelter este caracterizată prin sfioşenie, pasivitate, imaturitate şi dependenţa de aparţinători.
Confirmarea diagnosticului este realizată pe baza analizelor citogenetice şi a testărilor hormonale.
Investigaţia citogenetică necesară este analiza cromosomică. Principalele anomalii identificate sunt:
- trisomie XXY omogenă – 47,XXY (75% din cazuri)
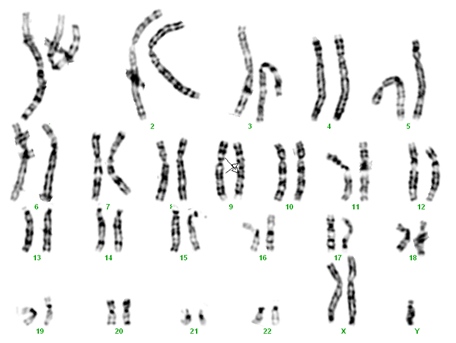
Trisomie XXY şi inversie pe cromosomul 9
- trisomie XXY în mozaic – 46,XY/47,XXY (~20% din cazuri)
- mozaicuri complexe de tipul 46,XY/47,XXY/48,XXXY sau 47,XXY/48,XXXY,
- polisomii XY omogene sau în mozaic de tipul: 48,XXYY, 48,XXXY sau 49,XXXXY.
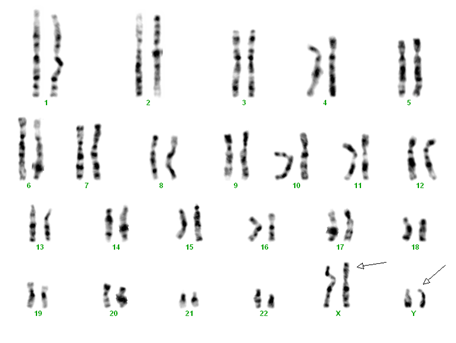
Tetrasomie XXYY
Analizele hormonale utile în diagnosticul sindromului Klinefelter sunt reprezentate de dozările de: testosteron – nivel scăzut, gonadotrofine hipofizare (FSH şi LH) – nivel crescut.
Tratament. Tratamentul se aplică numai postpubertar şi constă în administrare de hormoni sexuali masculini, cu scopul de inducere a dezvoltării caracterelor sexuale secundare masculine.
Tratamentul cu testosteron trebuie început de la vârsta normală de debut a pubertăţii (12-13 ani la băieţi) şi continuat, sub îndrumare endocrinologică pe tot parcursul vieţii active. În general terapia cu androgeni creşte masa musculară, determină îngroşarea vocii şi apariţia pilozităţii masculine, dar nu modifică aspectul şi funcţia testiculilor.
În general, ginecomastia nu răspunde la tratament hormonal, ci necesită tratament chirurgical, întrucât se însoţeste de creşterea riscului de cancer mamar.
O altă componentă a terapiei vizează tratamentul tulburărilor psihiatrice, care trebuie asigurat prin conlucrarea unui psihiatru şi a unui psiholog. Totuşi, de mare importanţă rămâne sprijinul familiei care permite ameliorarea integrării sociale şi creşte confortul psihic al pacientului.
Prognosticul. În majoritatea cazurilor, evoluţia pacienţilor cu sindrom Klinefelter este favorabilă. Astfel, la pacienţii cu trisomii XXY (omogene sau în mozaic) speranţa de viaţă este cvasinormală. În schimb, la indivizii cu polisomii XY (48,XXXY sau 49,XXXXY) speranţa de viaţă este limitată în condiţiile în care aceştia prezintă frecvent anomalii congenitale viscerale grave care pot determina deces în perioada copilăriei.
Inteligenţa este aproape normală, astfel încât integrarea socială a pacienţilor poate fi bună sau cel puţin acceptabilă. Totuşi, pot exista tulburări de vorbire care generează dificultăţi de adaptare socială. În plus, a fost remarcată o incidenţă crescută a tulburărilor psihiatrice de tip reactiv la factorii de stres.
Singurul aspect clinic care nu poate fi corijat este sterilitatea. În schimb, libidoul sexual nu este marcat sever, astfel încât activitatea sexuală este cvasinormală, mai ales dacă se administrează hormoni sexuali masculini.
Profilaxie. Deoarece sindromul Klinefelter determină sterilitate, boala nu se poate transmite la descendenţi (risc de recurenţă zero).
La cuplurile care au un copil cu SK, riscul de a avea un alt copil afectat este de obicei nesemnificativ (mai mic de 1%) dar este indicat un consult genetic înainte de o nouă sarcină, existând posibilităţi de diagnostic prenatal.
Depistarea bolii înainte de naştere necesită puncţie de vilozităţi corionice sau puncţie amniotică, urmată de efectuarea analizei cromosomice prin tehnici clasice sau prin metode de citogenetică moleculară (FISH).
Prof.dr. Eusebiu Vlad Gorduza